생물학 분야에서 AI 에이전트를 위한 길을 닦다
요약
AI 에이전트가 생물학 데이터 인프라를 더 안정적으로 활용하려면, 사람 중심의 복잡한 시스템을 넘어 확정적 데이터 검색 레이어 구축이 핵심이라는 연구 결과가 나왔어요.
인사이트
- 현재 AI 에이전트는 복잡하고 비정형적인 생물학 데이터 인프라(다양한 파일 형식, 분산된 데이터베이스, 수동 검색 스크립트 등)를 다루는 데 어려움을 겪고 있어요.
- `gget virus`와 같은 확정적 데이터 검색 레이어를 도입하면, AI 에이전트의 데이터 검색 정확도와 재현성이 거의 100% 가까이 향상되어, 모델 선택의 중요성이 줄어들고 더 저렴한 모델로도 신뢰성 높은 결과를 얻을 수 있어요.
- 미래에는 AI 에이전트의 능력이 향상되겠지만, 과학 연구의 신뢰성, 비용 효율성, 감사 가능성을 위해 '컨텍스트 엔진'과 같은 확정적이고 에이전트 친화적인 생물학 데이터 인프라를 구축하는 것이 여전히 중요해요.
왜 중요한가
AI 에이전트가 신약 개발, 질병 감시, 생물학 모델링 등 과학 분야에서 혁신을 이끌려면 정확하고 신뢰할 수 있는 데이터 접근이 필수적이에요. 이 연구는 현재 생물학 데이터 인프라의 한계를 명확히 보여주고, `gget virus` 같은 '확정적 검색 레이어'가 어떻게 이 간극을 메울 수 있는지 구체적인 해결책을 제시하고 있어요. 이는 과학 연구의 효율성과 신뢰성을 크게 높여, 궁극적으로 더 빠르고 정확한 과학적 발견을 가능하게 할 거예요.
앤트로픽 사이언스를 구독해 보세요
AI 기반의 발견, 실용적인 워크플로우, 그리고 과학 분야 전반의 현장 기록들을 담고 있어요.

이 글은 Laura Luebbert가 작성했어요. Ferdous Nasri, Sarah Gurev, Patrick Varilly, Krithik Ramesh, Nuala A. O’Leary, Jonah Cool, Bernhard Y. Renard, Pardis Sabeti, 그리고 Laura Luebbert의 연구를 바탕으로 하고 있어요.* 이 글에서 Laura Luebbert는 생물학 데이터 인프라를 에이전트 친화적으로 만들어야 한다고 주장하고 있어요. 한 사례 연구로, 그녀와 연구팀은 과학 연구 에이전트들(클로드, 바이옴니 오픈 소스(Biomni OSS) 1, 에디슨 분석 2, GPT)에게 NCBI 바이러스 데이터베이스에서 서열 데이터를 검색하도록 요청했어요. NCBI 바이러스는 바이러스 학자들이 감시나 진단 분석법 개발 같은 작업에 사용하는 데이터베이스예요. 그런데 아무리 강력한 모델들도 신뢰할 수 있는 데이터셋 구축에 필요한 수준의 정확도를 꾸준히 달성하지 못했어요. 하지만 그녀와 연구팀이 확정적 검색 레이어인 gget virus를 추가하자 정확도는 거의 100%까지 치솟았죠. 과학 에이전트에 대한 더 넓은 교훈은 확정적 검색 도구가 (현재로서는) 에이전트 워크플로우를 더 신뢰할 수 있게 만드는 데 매우 중요하다는 점이에요. 그리고 생물학 데이터베이스는 대규모 사용자로서 에이전트를 염두에 두고 설계될 필요가 있어요.*
AI 에이전트가 생물학 데이터 인프라를 탐색하게 하는 건 마치 자동차가 생기기 전에 설계된 오래된 도시를 운전하는 것과 비슷해요. 인프라 자체는 아름답고 심지어 사려 깊게 만들어졌을지 몰라도, 현대 차량이 다니기엔 너무 좁고 구불구불한 길이 가득하거든요 (독특한 파일 형식, 여기저기 흩어진 데이터베이스, 일회성 검색 스크립트 등).
그래서 코딩 에이전트들이 생물학 에이전트보다 훨씬 빠르게 발전했어요. 소프트웨어는 보통 구조화된 디지털 워크플로우와 안정적인 인터페이스를 제공하지만, 데이터 검색 및 검증에 필요한 계산 생물학 인프라는 종종 취약하고, 이질적이며, 특정 프로세스에 의존적이거든요. 이런 인프라를 다루는 도구들은 특정 영역이나 가설에 맞춰 맞춤 제작될 수밖에 없어요. 게다가 소프트웨어는 빠르게 컴파일하고 검증할 수 있는 테스트 가능한 결과물(예: 프로젝트 테스트를 통과하는 패치를 생성해서 GitHub 이슈를 해결하는 것)을 제공하는 반면, 생물학 분야에서는 간단하고 검증 가능하면서도 의미 있는 결과물을 얻기 어려워요.
결국 생물학 에이전트의 병목 현상은 추론 능력뿐 아니라 생물학 데이터 쿼리를 위한 확정적 실행 레이어가 널리 보급되지 않았다는 점에 있어요. 과학자는 자신의 의도(예: 이 도메인을 가진 모든 인간 키나아제를 찾아 구조를 가져와 줘)를 표현할 수 있지만, 에이전트는 필요한 정보를 담고 있는 데이터베이스에 안정적으로 접근할 방법이 없는 경우가 많아요.
생물학 및 과학 워크플로우에서는 작은 실수라도 심각한 결과를 초래할 수 있어요. 예를 들어, 잘못된 유전체 빌드에서 좌표를 가져오면 후속 생물학적 해석이 무효화될 수 있죠. 의도치 않게 RefSeq와 GenBank 기록을 섞거나, 부분 유전체를 완전한 유전체로 간주하거나, 분절된 바이러스에서 세그먼트 이름을 혼동하거나, 일관성 없는 메타데이터 필드 때문에 관련 기록을 놓치는 것들도 마찬가지예요. 연구의 아름다움이자 어려운 점은 바로 세부 사항들이 종종 치명적으로 중요하다는 점이에요.
이탈리아의 언덕 마을을 운전하는 것과 비슷해요. 길이 너무 좁고, 코너가 너무 날카로우며, 경로가 현지 지식에 따라 달라진다면 아무리 강력한 자동차라도 소용이 없잖아요. 에이전트가 전염병 대응부터 약물 설계, 생물학 모델링에 이르기까지 과학적 발견을 돕기를 원한다면, 에이전트가 사람만큼이나 안정적으로 탐색할 수 있는 생물학 데이터 인프라를 구축해야 해요.
에이전트의 필요와 인간이 만든 도구 사이의 이런 불일치는 생물학에만 국한된 건 아니에요. 오직 사람만을 위해 설계된 환경에 에이전트가 투입될 때마다 비슷한 마찰이 발생하죠.
몇 달 전 Andrej Karpathy는 AI 시대의 소프트웨어에 대한 강연을 했는데, 너무나 익숙하게 들리는 불평을 늘어놓았어요. 그는 작은 웹 앱을 뚝딱 만들었지만, 실제로 서비스화(인증, 결제, 배포)하려 하자 브라우저 대시보드를 이리저리 클릭하느라 일주일을 허비했대요.
그가 요약했듯이, "코딩이 제일 쉬웠어요! 대부분의 작업은 브라우저에서 이것저것 클릭하는 거였죠." 문서에는 계속 "이 URL로 가서, 이 드롭다운을 클릭하세요"라고 나와 있었대요. 그의 결론은 아무도 이런 일을 할 필요가 없어야 한다는 거였어요. 대신, 우리는 에이전트를 위해 구축해야 해요.**
카르파티는 소프트웨어 에이전트 세계에서 새로운 경험을 했지만, 생물학 연구자들은 오랫동안 똑같은 문제로 씨름해 왔어요. 바로 이질적인 정보, 암묵적인 규칙, 그리고 브라우저를 클릭하는 인간을 중심으로 구축된 환경에서 지능형 시스템을 작동시키려는 고통이죠.
AI 에이전트가 등장하기 훨씬 전부터 계산 생물학자들과 유전학자들은 이미 이 문제를 해결하기 위해 전통적인 계산 생물학 도구들을 만들어내기 시작했어요. 바이오파이썬(Biopython), 바이오펄(BioPerl), 바이오줄리아(BioJulia), 엔트레즈 다이렉트(Entrez Direct), 바이오마트(BioMart), gget 같은 패키지와 수많은 다른 워크플로우 라이브러리들이 모두 생물학 데이터를 브라우저 인터페이스 밖으로 꺼내 연구자들이 직접 계산할 수 있는 곳으로 옮기려는 노력의 일환이었어요.
문제는 생물학 데이터가 단일 인터페이스를 가진 단일 데이터베이스에 존재하는 게 아니라는 점이에요. 각기 다른 식별자, 규칙, 형식, 필터링 로직, 그리고 프로그래밍적 접근 수준을 가진 복잡한 도로망과 같죠. 일부 데이터는 프로그래밍 방식으로 쉽게 접근할 수 있지만, 다른 것들은 그렇지 않아요.
특히 바이러스학 분야는 어려운 사례 중 하나예요. 백신 및 진단 분석법 설계부터 단백질 모델 학습 데이터 구축에 이르는 연구 워크플로우는 종종 NCBI 바이러스에서 서열을 검색하는 것으로 시작해요. NCBI 바이러스는 젠뱅크(GenBank), RefSeq, 그리고 Pathoplexus를 포함한 국제 INSDC 생태계의 바이러스 서열 기록들을 검색 가능한 웹 인터페이스 뒤에 모아둔 곳이에요. 바이러스 발병 감시 도구를 개발하는 연구자들은 이런 검색 과정 뒤에 얼마나 많은 전문가 지식이 숨어있는지 직접 경험으로 알고 있어요. 바이러스학 연구실에서는 NCBI 바이러스 데이터셋 큐레이션 지침이 종종 사용자가 웹 인터페이스에서 수동으로 재현해야 하는 복잡한 필터의 긴 목록으로 공유되곤 해요. 바로 카르파티가 불평했던 그런 브라우저 클릭 워크플로우죠.
현재 콩고민주공화국에서 분디부교 바이러스(Bundibugyo virus)로 인한 에볼라병 발병은 간소화된 바이러스 데이터 접근이 현실에서 생사를 가르는 결과를 초래할 수 있음을 보여주는 극명한 사례예요. 2026년 5월 14일, 콩고민주공화국의 INRB 킨샤사는 13개의 혈액 샘플을 분석했고, 다음 날 8건에서 분디부교 바이러스병을 확인했어요. 4 이후 에볼라 발병이 선언되었죠. 5월 29일까지 WHO는 콩고민주공화국에서 200명 이상의 사망자를 포함해 1,000건 이상의 확진 및 의심 사례를 보고했어요. 연구자들은 또한 첫 번째 거의 완전한 발병 유전체를 생성하여, 이 발병이 새로운 유출 사건으로 인해 발생했음을 확립하는 데 도움을 주었어요.
이 유전체들은 공중 보건 관계자들에게 세 가지 시급한 질문을 던져요. 첫째, 이 발병 바이러스는 이전에 발견된 에볼라 바이러스와 얼마나 다른가요? 둘째, 기존 진단법으로 여전히 이 바이러스를 감지할 수 있을까요? 셋째, 기존 치료법으로 여전히 이 바이러스로부터 보호받을 수 있을까요? 이 질문들에 답하려면 새로운 유전체를 NCBI 바이러스와 Pathoplexus(NCBI 바이러스와 동기화됨)를 통해 사용 가능한 기존 에볼라 유전체와 비교해야 해요. 하지만 이 과정은 쉽게 자동화되지 않고, 이 분석의 첫 단계는 웹 인터페이스를 수동으로 클릭하고, 복잡한 필터를 손으로 재현하고, 결과 데이터셋이 완전하고 정확하기를 바라는 작업으로 이루어져요.
이 워크플로우가 자동화하기 어려운 이유는 NCBI 바이러스의 필터링 로직 대부분이 오직 이 웹 인터페이스에만 존재하기 때문이에요. 이건 사람에게도 번거로운 일이고, 에이전트에게는 최악이죠. 만약 연구자가 2025년에 발표된, 표면 당단백질을 포함하는 모든 SARS-CoV-2 서열을 원한다면, 숙련된 바이러스 학자는 브라우저에서 몇 번의 클릭만으로 가능할 거예요. 하지만 프로그램적으로는 여러 API(REST, Datasets, E-utilities)를 연결하고, 페이지별로 결과를 가져오고, 식별자를 조정하고, 수백 기가바이트의 데이터를 다운로드한 다음 로컬 필터링 후 대부분을 버려야 하는 수백 줄짜리 스크립트가 필요할 수 있어요.
어떤 리소스에 API가 있더라도, 에이전트가 안정적으로 사용하기 어려운 여러 가지 이유가 있어요. 예를 들어, API가 웹 인터페이스와 동일한 필터링 의미를 노출하지 않거나, 메타데이터 필드가 제대로 문서화되어 있지 않거나 일관성 없이 표준화되어 있거나, 출처에 따라 식별자가 변경되거나, 또는 '올바른 답'이 전문가인 인간은 알지만 기계는 추론해야 하는 규칙에 의존하는 경우 등이 그렇죠.
에이전트와 데이터베이스를 연결하는 과제를 더 잘 이해하기 위해, Anthropic 팀은 최첨단 과학 연구 에이전트들(클로드, 바이옴니 OSS, 에디슨 분석, GPT)이 현재 사용 가능한 인프라를 사용해서 NCBI 바이러스에서 바이러스 서열을 검색할 때 얼마나 유능한지 테스트하기 위한 벤치마크를 개발했어요. 이 벤치마크인 VirBench에는 40가지 병원체를 아우르는 120개의 실제 바이러스 서열 쿼리가 포함되어 있으며, 수동으로 검증된 실제 정답 수치를 가지고 있어요. 쿼리들은 바이러스 감시, 진단 분석법 설계, 단백질 모델 학습 데이터 구축에서 나타나는 작업들을 반영하고 있죠. 예를 들어, 한 쿼리는 에이전트에게 “다음 기준을 준수하는 TaxID 3052462 (Orthoebolavirus zairense (ZEBOV))에 대한 NCBI의 바이러스 서열을 가져오세요: 숙주 유기체: 인간, 샘플 채취 지리적 위치: 아프리카, 채취일: 2014년 1월 1일 이후 및 2014년 6월 20일 이전, 최소 서열 길이: 15,200 염기, 최대 모호한 문자(N) 수: 1,900개, 실험실 계대 배양 샘플 제외”라고 요청했어요.
에이전트들이 스스로 이 쿼리들을 해결하도록 맡겼을 때, 성능은 시스템마다 크게 달랐고, 최신 프론티어 모델에서는 상당히 개선된 모습을 보였어요. 하지만 아무리 강력한 모델들도 신뢰할 수 있는 데이터셋 구축에 필요한 수준의 정확도와 재현성을 꾸준히 달성하지 못했죠. 클로드 소네트 4(Claude Sonnet 4), 클로드 오푸스 4.7(Claude Opus 4.7), 바이옴니 OSS, 에디슨 분석, GPT-5.2-pro, 그리고 GPT-5.55는 16.9%에서 91.3%에 이르는 평균 정확도를 기록했어요. 이런 데이터 검색 작업에서는 사실상 100%의 정확도가 요구돼요. 어떤 경우에는 누락되거나 잘못된 기록 하나가 진단 분석법이 현재 유행하는 다양성을 커버하는지 여부나, 발병이 실제보다 몇 주 빠르거나 늦게 시작되었다고 추론되는지를 결정할 수 있거든요. 게다가 같은 모델이 동일한 질문을 세 번 받았을 때도 상당히 다른 답변을 내놓는 경우가 많았는데, 이는 신뢰할 수 있는 과학 워크플로우에 필요한 정확성과 재현성을 모두 해치는 결과였어요. 위에 언급된 에볼라바이러스 쿼리 사례의 경우, 소네트 46은 매번 동일한 프롬프트를 받았음에도 불구하고, 첫 번째 실행에서는 106개 서열(예상: 266개), 두 번째 실행에서는 15개, 세 번째 실행에서는 5개를 반환했어요.
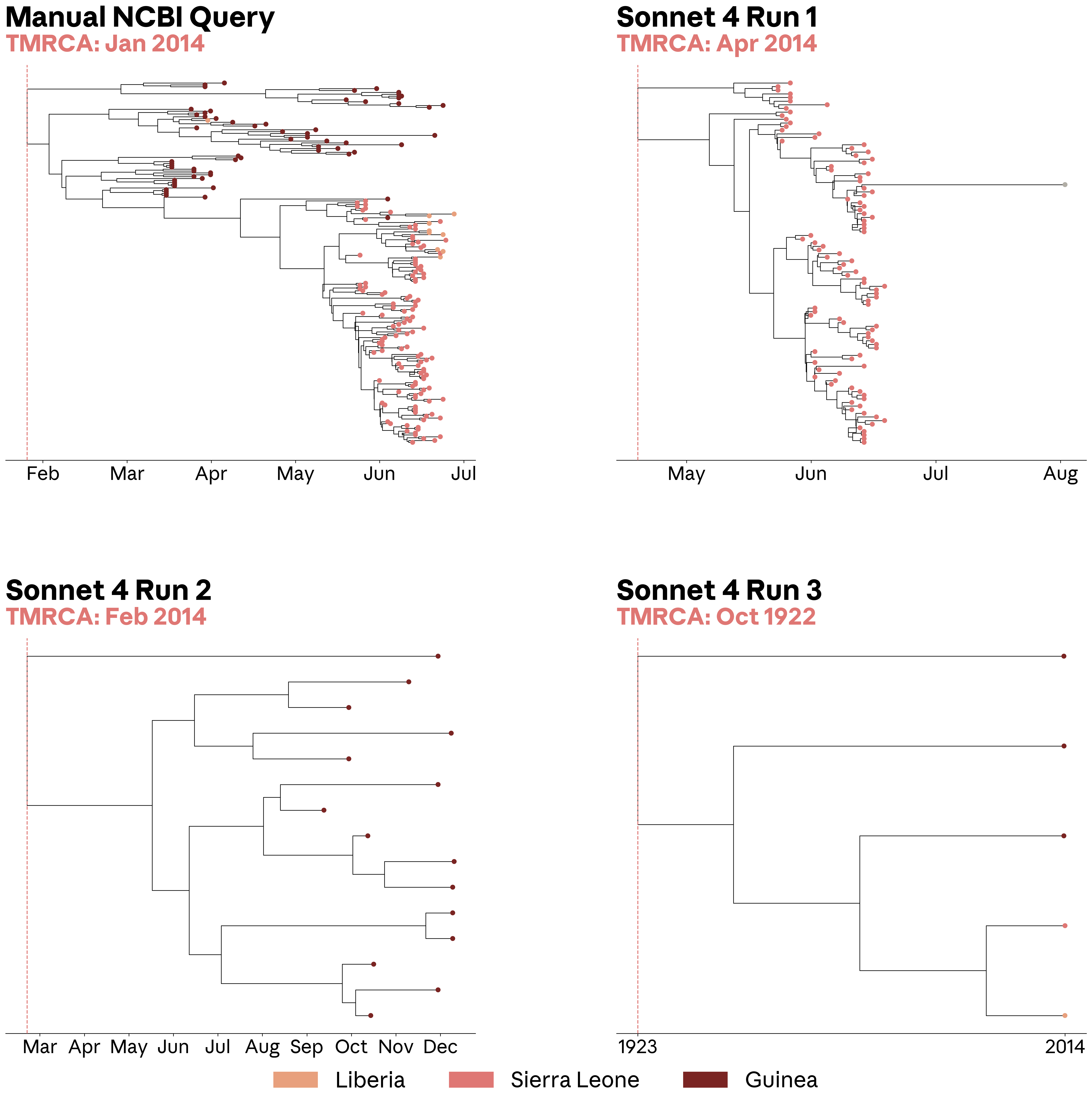
이런 불일치는 후속 분석에 영향을 미쳐요. 연구팀은 위에 제시된 쿼리를 사용해서 에볼라바이러스 서열을 검색하고 계통발생학적 트리(phylogenetic tree)를 구축했어요. 이는 발병 중에 바이러스 샘플이 어떻게 관련되어 있는지 재구성하는 표준 분석법이죠. 계통발생학적 트리에서 얻을 수 있는 중요한 양 중 하나는 최근 공통 조상까지의 시간(TMRCA) 추정치예요. 이것은 발병의 추론된 기원 날짜이며, 바이러스가 언제 어디서 시작되었는지, 그리고 얼마나 오랫동안 유행했는지에 대한 결론을 바꿀 수 있어요. 이 경우, 수동으로 큐레이션된 NCBI 바이러스 서열 세트로 구축된 트리는 2014년 1월 TMRCA를 복구했는데, 이는 2014년 에볼라바이러스 발병에 대한 이전 보고서 (95% 최고 사후 밀도 범위는 1월 27일부터 3월 14일)와 일치했어요. 반면, 소네트 4가 검색한 세 가지 서열 세트 중 두 개는 눈에 띄게 불완전했는데, 하나는 추론된 TMRCA를 1922년으로 되돌려 놓은 트리였어요. 나머지 데이터셋(실행 1)은 겉보기에는 그럴듯했지만, 기니(Guinea) 지역의 서열을 검색하지 못했고 추정 TMRCA를 2014년 4월로 변경하여 발병의 추론된 시점을 바꾸어 버렸어요.
NCBI 바이러스 검색 시도 간의 가변성은 치료법에 대한 결론에도 영향을 미칠 수 있어요. Anthropic 팀은 에볼라바이러스 당단백질 서열을 검색해서 현재 진행 중인 에볼라바이러스 발병에서 자이르 에볼라바이러스에 대해 개발된 항체 치료제이자 WHO 우선 치료 후보인 마프티비맙(maftivimab)과 MBP134가 결합하는 에피토프를 조사했어요. 연구팀은 관련 자이르 에볼라바이러스 서열에서 이 항체들이 표적으로 삼는 영역에 이전에 돌연변이가 발생했는지 여부를 질문했죠. 이런 종류의 분석은 연구자들이 바이러스가 진화함에 따라 치료법이 계속해서 환자를 보호할 수 있을지에 대한 감을 얻는 데 도움을 줄 수 있어요. 만약 기본 서열이 불완전하거나 잘못 가져와진다면, 결론을 완전히 뒤집어놓을 수 있거든요. Anthropic 팀의 예시에서, 소네트 4가 검색한 서열은 첫 시도에서 수동 NCBI 쿼리를 통해 얻은 결과와 거의 일치했어요. 하지만 반복 실행에서는 대부분의 돌연변이 잔기를 놓쳤고요. 그리고 세 번째 실행에서는 다른 잔기 세트를 강조하여, 이 표적 영역의 변동성에 대해 세 가지 다른 인상을 주었어요.7
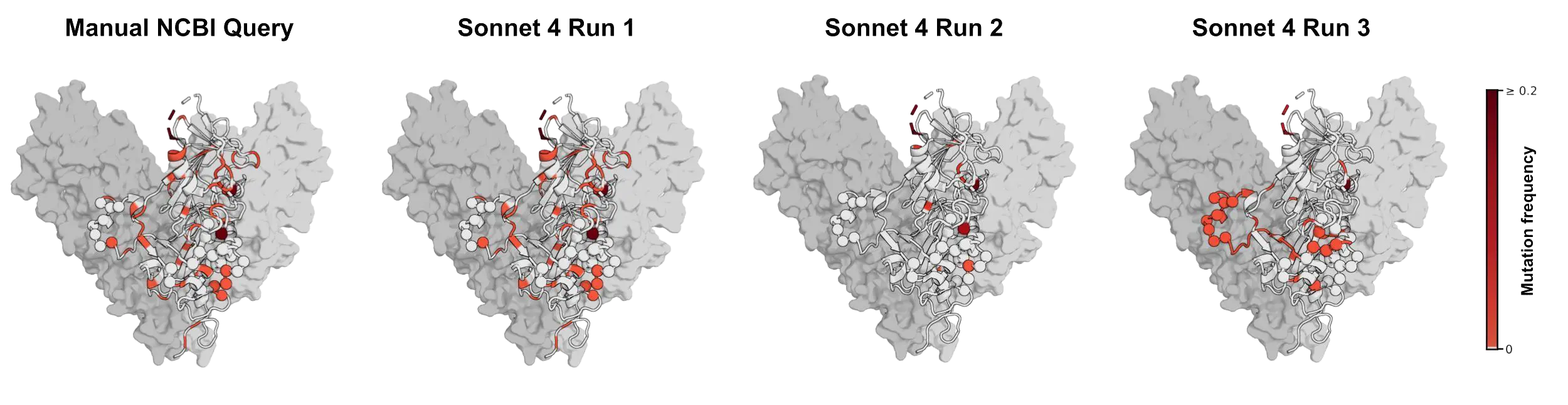
이 두 가지 사례 모두 과학 분야의 더 넓은 패턴을 보여줘요. 사소해 보이는 검색 선택의 세부 사항이 생물학적 결론을 바꿀 수 있다는 점이죠. 이 경우, 바이러스 서열 검색에서 모델의 일관성 없는 성능과 오류 발생 방식은 대부분의 변동이 인프라의 한계 때문이라는 것을 보여주었어요. 에이전트는 대규모 결과 세트를 검색하지 못했을 때 과소 집계했고, 필터가 잘못 적용되었을 때 과대 집계했어요. 예를 들어, 인플루엔자 A, HIV-1, SARS-CoV-2처럼 사용 가능한 기록이 많은 바이러스의 경우, 검색 도중에 중단하거나 후속 필터링이 잘못되면 최종 데이터셋을 크게 왜곡할 수 있어서 예상 수치에서 가장 큰 편차가 발생했어요. 또한 맥락, 규칙 또는 정보가 저장된 위치에 따라 의미가 달라지는 메타데이터 필드에서도 어려움을 겪었고요. 쿼리가 복잡해질수록, 특히 세네 개 이상의 필터가 동시에 적용될 때 성능이 저하되었어요.
결론적으로 에이전트들은 종종 작업을 시도할 만큼 충분히 잘 이해했지만, 이를 수행하고, 검증하고, 반복할 수 있는 기계가 실행 가능한 방법이 부족했어요. 결과로 나온 답변은 그럴듯해 보이지만 여전히 틀릴 수 있었는데, 이는 서열 검색이 보통 훨씬 긴 생물학적 워크플로우의 첫 단계라는 점에서 특히 위험한 일이에요.
VirBench와 gget virus에 대한 더 자세한 설명을 보려면 프리프린트를 읽어보세요.
바이러스 데이터 검색을 에이전트와 사람이 직접 호출할 수 있는 형태로 바꾸기 위해, 연구팀은 NCBI 연구원들과 협력해서 gget virus를 개발했어요. 처음에는 올바른 API 호출에 연결하는 간단한 문제처럼 보였죠. 하지만 실제로는 훨씬 어려웠어요. NCBI 바이러스는 미국, 유럽, 일본 전역에 걸쳐 국제적으로 동기화되는 서열 데이터베이스를 포함한 여러 기본 리소스들을 아우르는 포털이거든요. 그래서 겉으로는 간단해 보이는 쿼리에도 종종 여러 곳에서 정보를 조합해야 했어요.
NCBI 바이러스 웹 인터페이스의 동작을 재현하기 위해, gget virus는 REST, Datasets, E-utilities API를 포함한 여러 하위 시스템들을 조율해야 해요. gget virus는 기존 API를 통해 적용할 수 있는 필터와, 웹 인터페이스가 단일 프로그래밍 엔드포인트에서는 사용할 수 없는 필터링 동작을 노출하기 때문에 로컬에서 확인해야 하는 필터를 결정해요. SARS-CoV-2 및 인플루엔자 A 데이터셋과 같은 대규모 결과 세트가 임의로 잘리지 않고 포괄적으로 검색되도록 배치 처리를 담당하죠. 필터링이 특정 바이러스 단백질을 포함하는지 여부를 나타내는 GenBank 기록과 같이 별도의 데이터베이스에 저장된 추가 정보에 의존하는 경우, gget virus는 해당 기록을 검색하고, 이를 사용하여 필터를 적용하며, 관련 GenBank 정보를 최종 결과에 보존해요. 그런 다음 사람과 기계 모두 읽을 수 있는 표준화된 출력과 함께 최종 결과가 어떻게 생성되었는지 보여주는 상세 로그를 반환합니다.8
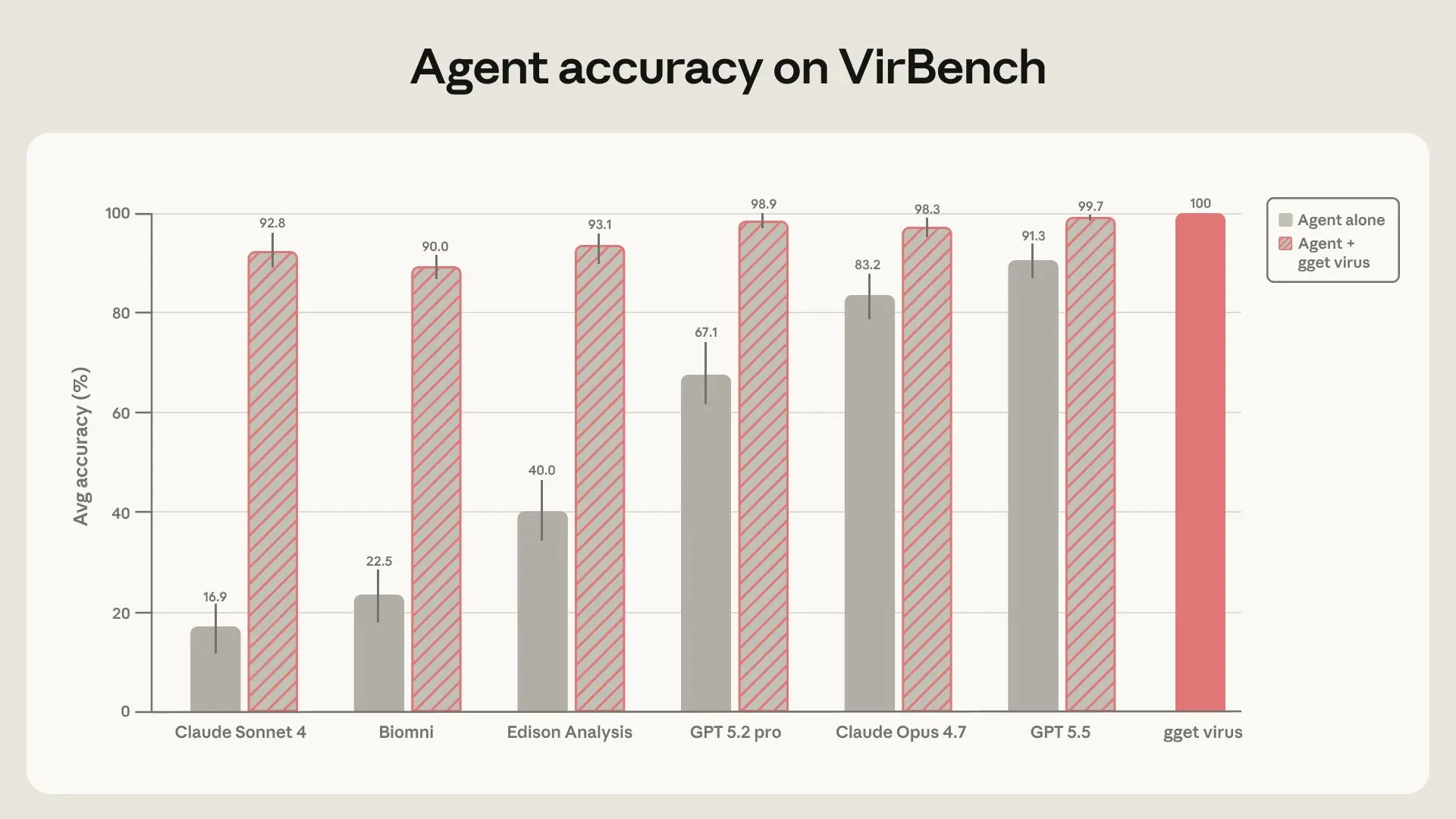
연구팀이 에이전트들에게 gget virus에 대한 접근 권한을 주자, 모든 에이전트의 정확도가 90% 이상으로 올라갔고, GPT-5.5의 경우 99.7%에 달했어요. 실행 간 가변성은 거의 사라졌고, 모델 간 성능 격차도 dramatically 줄어들었죠. 다시 말해, 확정적 검색 레이어를 추가함으로써 모델 선택의 중요성이 훨씬 낮아졌다는 거예요. 이는 신뢰할 수 있는 데이터셋 구축이 최신 또는 가장 비싼 모델에 대한 접근이나 특정 데이터베이스에 가장 적합한 모델을 아는 것에 의존해서는 안 된다는 점을 고려할 때 특히 중요해요. 대신, 적절한 도구와 결합된 저렴한 모델이 가변성을 줄이고 더 넓은 접근성을 가능하게 합니다.
gget virus는 복잡하고 브라우저 기반인 검색 워크플로우를 정확하고 재현 가능한 인터페이스로 변환해서 기존 에이전트의 바이러스 데이터 검색 신뢰도를 높여줘요. 보행자 친화적인 도시 비유로 돌아가 보면, 보행자 인프라 밑에 고속도로 터널을 추가한 것과 같아요. 진입로와 진출로, 원활한 인터체인지, 그리고 알려진 마일 스톤에 연결된 출구 번호까지 갖춘 그런 터널 말이죠.
우리는 모델이 가설을 생성하거나, 실험을 설계하거나, 메커니즘을 추론할 때 창의적이기를 원해요. 하지만 그 창의성 아래에 있는 레이어, 즉 유전자 식별자, 스키마, 검색 로직, 좌표계, 메타데이터 규칙, 데이터 접근 경로는 지루할 만큼 안정적이어야 해요 (다시 말해, 확정적이어야 하죠). gget virus는 이런 컨텍스트 엔진, 즉 생물학 데이터를 위한 안정적이고 에이전트가 접근 가능한 인프라를 구축하려는 더 넓은 노력의 한 예시예요. 다른 노력들도 AI for Science 시스템에서 나오고 있는데, 이들 중 다수는 ToolUniverse, 에디슨 사이언티픽의 로빈(Robin), 바이옴니(Biomni), 그리고 관련 생물 의학 에이전트들을 포함해 에이전트를 생물학 데이터 소스에 연결하는 모델 하네스에 의존하고 있어요. 과제는 그런 확정성이 어디에 속해야 하고, 어떻게 구축해야 하는지 알아내는 것이에요.
모델 역량이 얼마나 빠르게 변화하는지 고려하면 커넥터와 하네스에 대한 작업은 더욱 복잡해져요. 위 결과에서 모델 곡선을 미래로 연장해 보면, gget virus 같은 도구의 이점이 거의 0에 수렴하는 (아주 가까운) 미래를 상상하기 쉽죠. 에이전트가 복잡한 포털을 탐색하고, 식별자를 조정하고, 페이지 매김을 올바르게 처리하며, 스스로 오류에서 복구할 만큼 충분히 똑똑해지는 거죠. 하지만 에이전트가 그 일을 할 수 있다고 해도, 그 작업이 매번 에이전트에 의해 처리(그리고 재발명)되어야 한다는 뜻은 아니에요. 혼란스러운 생물 정보학 워크플로우를 뚫고 나갈 수 있는 모델이라도, 일상적인 과학 작업에는 여전히 너무 비싸고, 너무 느리며, 감사하기 어렵거나, 신뢰하기 어려울 수 있어요. 그리고 에이전트가 결국 오늘날의 하네스를 쓸모없게 만든다고 해도, 생물학 데이터베이스에 대한 교훈은 여전히 유효해요. 우리는 사용자를 생각할 때 에이전트를 염두에 두어야 하고, 확장을 고려해서 구축해야 한다는 거죠.
이 글을 개선하는 데 도움이 된 사려 깊은 피드백, 꼼꼼한 편집, 그리고 유익한 대화를 제공해 준 Xander Balwit, Ethan Dyer, Stuart Ritchie, Rebecca Hiscott, Alyssa Morrow, Keir Bradwell, Eric Kauderer-Abrams, Jonah Cool, Andrej Karpathy, Patrick Varilly, Cesar Arze, Blake Lash, Philine Guckelberger, Nisha Gopal, Elliot Hershberg, Pardis Sabeti, 그리고 Jonathan Feldman에게 감사드립니다.
특히 예시 바이러스학 분석을 개발하고 수행하는 데 도움을 준 Sarah Gurev와 Gage Moreno, 그리고 이 글의 아이디어, 구성, 작성에 상당한 기여를 해준 Ferdous Nasri와 Krithik Ramesh에게 특별히 감사드립니다.
AI 기반의 발견, 실용적인 워크플로우, 그리고 과학 분야 전반의 현장 기록들을 담고 있어요.